Modeling cell metabolism and protein expression
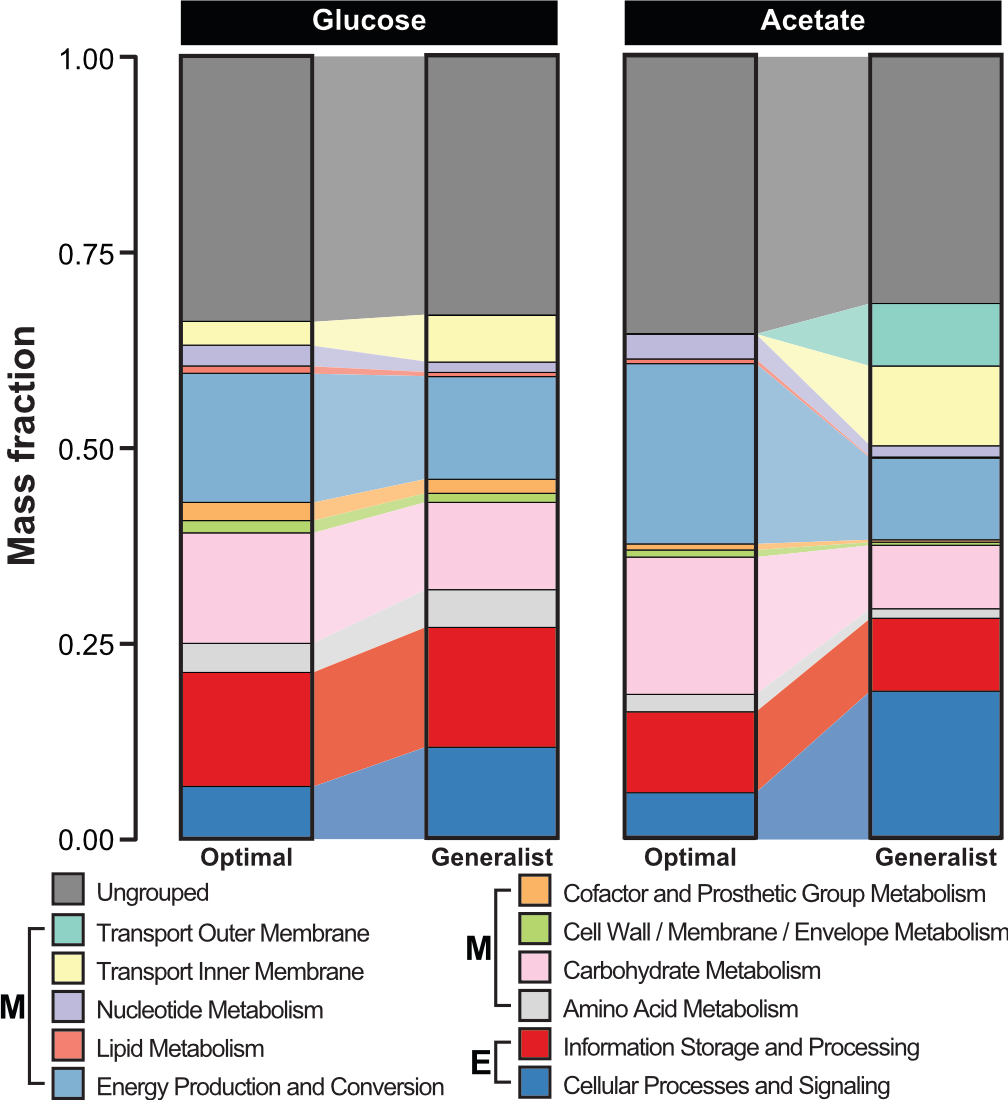
A By imposing data-driven constraints onto ME models, we can more accurately predict phenotypes that reflect the generalist life-style of microbes like E. coli.
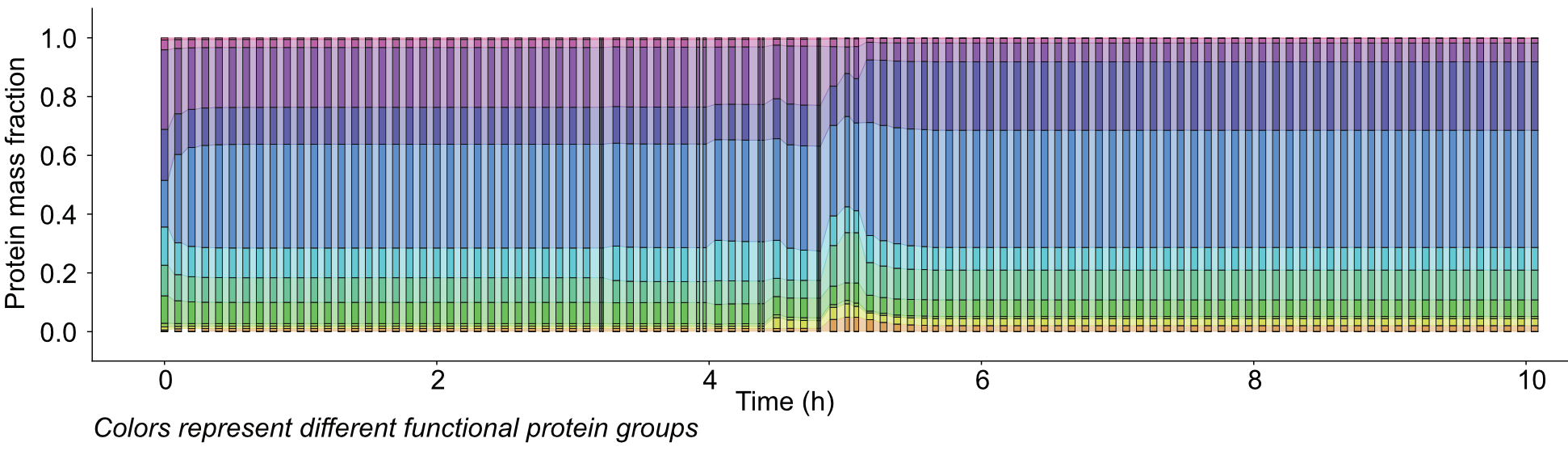
B Limitations in protein expression machinery constrains how quickly the proteome can be reallocated under dynamic environmental changes. By expressing this constraint mathematically, our models more accurately predict protein expression dynamics, including shifts in energy metabolism (shifting reliance from oxidative phosphorylation to substrate-level phosphorylation) under dynamically changing nutrient availability.
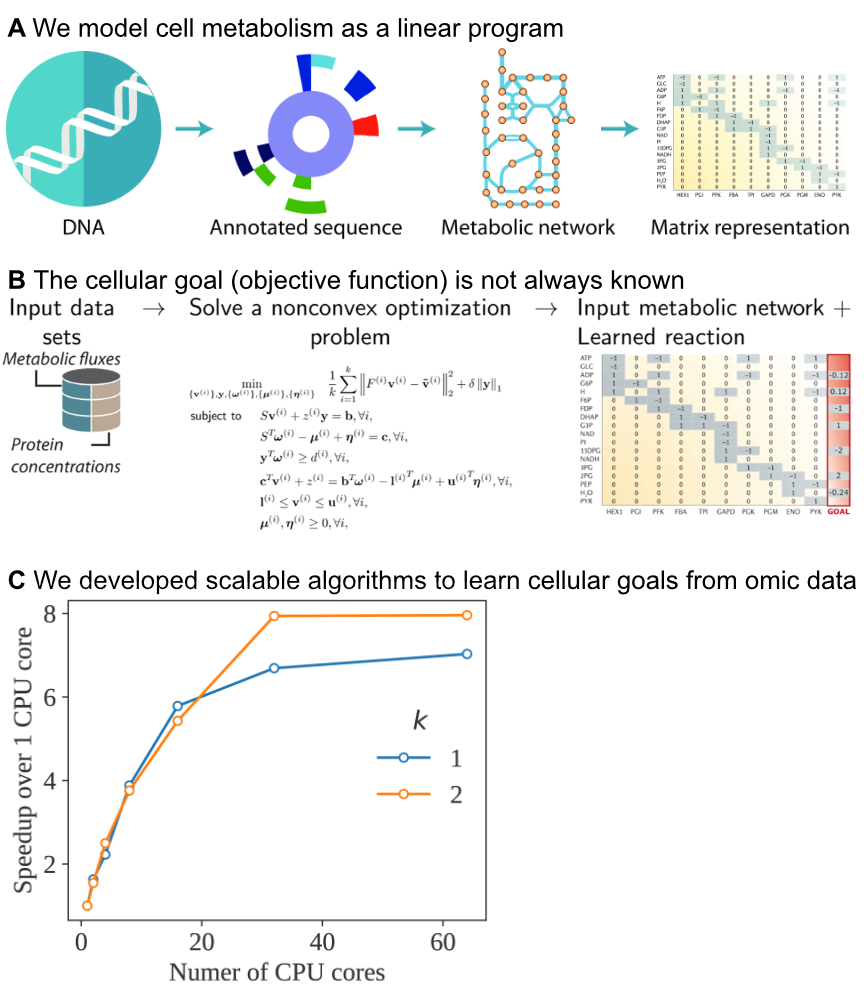
Genome-Scale Modeling
Cell metabolism is the set of chemical reactions that sustain life for an organism. Even the simplest microbes possess a complex metabolism comprised of a network of hundreds of enzyme-catalyzed reactions.
Given the genome sequence of an organism, systems biologists can reconstruct the metabolic reaction network. This network is formulated as a matrix, where rows represent metabolites and columns represent reactions.
To predict the reaction rates (fluxes) throughout this network, we use flux balance analysis (FBA) (Orth et al., 2011). FBA was developed in the early 90s and in numerous studies since, FBA has accurately predicted systems-level metabolic responses in the context of biotechnology, infectious disease, cancer metabolism, and environment engineering.
We employ an extension of FBA developed since 2012: metabolism and macromolecule expression (ME) models. ME models are considerably more detailed reconstructions that comprise metabolism, protein expression machinery, protein complex formation, (metal) cofactor incorporation, sigma factor networks, protein translocation, and additional mechanisms for specific organisms.
Scalable Algorithms for Genome-Scale Modeling
The cellular goal can be challenging to define for many organisms, including human tissue, microbial pathogens, and cancer cells.
A promising approach is to estimate these goals directly from omics measurements, given a starting metabolic reconstruction. A particuarly flexible method is estimating new linear constraints that model unknown biochemical reactions that constrain the cell's operation.
However, this approach requires solving a nonconvex optimization problem, which may not scale to large models. To tackle this challenge, we develop scalable algorithms using distributed computing on CPUs and GPUs. Our algorithms thus learn new models from high-throughput data sets, leading to increasingly accurate prediction of cellular behavior under conditions that were previously difficult to model.
Researchers will have ample opportunity to deploy machine learning and distributed algorithms on big biological data sets. These algorithms can improve the accuracy of model predictions, or to help understand biological mechanisms by constructing explainable models from data.
Our lab aims to:
- develop scalable algorithms to estimate model parameters from multi-omics data (i.e., data sets comprised of multiple omics technologies)
- learn models of metabolism and protein expression from multi-omics data, including microbial community models and host-pathogen models
Researchers

Laurence Yang
Assistant Professor

Jiao Zhao
Research Associate

Sanjeev Dahal
Postdoctoral Fellow

Herbert Yao
MASc Student

Xinran Li
MASc Student
- Publications
- Pseudomonas aeruginosa (preprint)
- Cell goals paper
- DynamicME paper
- Proteome constraints
- Software
- DynamicME code
- Cellgoal code
- Academic Collaborators
- Jose Bento
- Funding
- 2020 - : NSERC Discovery Grant